

Translate this page into:
Management of Life-threatening Refractory Chylothorax and Chylopericardium due to Lymphangiomatosis

Corresponding Author: Hussam K. Hamadah, Section of Pediatric Cardiac ICU, King Abdulaziz Cardiac Center, King Abdulaziz Medical City, Ministry of National Guard–Health Affairs, Mail Code: 1423, P.O Box 22490, Riyadh 11426 Kingdom of Saudi Arabia. E-mail: hamadahmo@ngha.med.sa
-
Received: ,
Accepted: ,
How to cite this article: Hamadah HK, Kabbani MS. Management of Life-threatening Refractory Chylothorax and Chylopericardium due to Lymphangiomatosis. Am J Sonogr 2018, 1(2) 1-4.
Abstract
Lymphangiomatosis is a rare disorder. Chylous effusion and osteolytic lesions are diagnostic clues. Radiographic findings can be suggestive of the disease, but the final confirmatory diagnosis is made histologically. We report a case of an infant who presented with life-threatening refractory chylothorax and chylopericardium due to diffuse pulmonary lymphangiomatosis, leading to critical condition with challenging management. With multidisciplinary approach, the child improved and was discharged home with satisfactory outcome.
Keywords
Chylopericardium
Chylothorax
Lymphangiomatosis
Interleukin-6
Pegylated interferon alfa-2b

INTRODUCTION
Lymphangiomatosis is a rare disease in children. It involves lungs, bones, pleura, and other viscera in a diffuse fashion.[1-3] The lungs are the main affected organ and their involvement determines prognosis.[1-3] The disease has no specific presentation and patients may develop non-specific symptoms such as dyspnea, cough, and wheezing mimicking other lung diseases or may present with symptoms related to pleural or mediastinal fluid accumulation. The disease is believed to be congenital with poor prognosis when complicated by chylothorax and chylopericardium.[1-3] Diffuse lymphangiomatosis management is controversial due to the rarity of this disease and lack of knowledge and experience with such cases.[1-3]
The clinical presentation, radiological findings, histological features, and approach to treatment are discussed in this case report.
CASE REPORT
A 20-month-old boy previously healthy with unremarkable past medical history presented to emergency room with impending respiratory failure and shock related to significant right pleural effusion and pericardial tamponade (Graph 1). His family history and past medical history were non-contributory except for a presumed diagnosis of bronchiolitis 3 weeks prior to this admission.
![A 20-month-old boy with diffuse lymphangiomatosis who presented with chylothorax and chylopericardium; effect of therapy with pegylated interferon alfa-2b and other medications on drainage volume with correlated images (chest X-ray [CXR] and echocardiography). (a) CXR on presentation revealed cardiomegaly with the right pleural effusion, and echocardiography apical four chambers view is consistent with early signs of tamponade. (b) CXR and subcostal echocardiography view revealed significant effusion in the pleural and pericardial space in spite of drains. The decline in drainage volume before pericardial window and pleuroperitoneal shunt was due to frequent drains obstructions and not due to clinical improvement. (c) CXR and subcostal echocardiography view after pericardial window and pleuroperitoneal shunt revealed no further effusion. (d) CXR and apical four chambers echocardiography on discharge revealed no effusion after removal of chest tubes in the presence only of pleuroperitoneal shunt.](/content/3/2018/1/1/img/AJS-1-2-g001.png)
- A 20-month-old boy with diffuse lymphangiomatosis who presented with chylothorax and chylopericardium; effect of therapy with pegylated interferon alfa-2b and other medications on drainage volume with correlated images (chest X-ray [CXR] and echocardiography). (a) CXR on presentation revealed cardiomegaly with the right pleural effusion, and echocardiography apical four chambers view is consistent with early signs of tamponade. (b) CXR and subcostal echocardiography view revealed significant effusion in the pleural and pericardial space in spite of drains. The decline in drainage volume before pericardial window and pleuroperitoneal shunt was due to frequent drains obstructions and not due to clinical improvement. (c) CXR and subcostal echocardiography view after pericardial window and pleuroperitoneal shunt revealed no further effusion. (d) CXR and apical four chambers echocardiography on discharge revealed no effusion after removal of chest tubes in the presence only of pleuroperitoneal shunt.
The patient required urgent pericardiocentesis and right pleural thoracentesis after initial resuscitation and airway stabilization. 1 L of pericardial and 1 L of pleural fluid were drained. Analysis and cultures of drained fluid revealed sterile exudative chylous fluid that was negative for malignancy (white cell count of 5300 per mm3 with a lymphocyte fraction of 83%, fluid triglyceride 2.19/serum 1.02 mmol/L, fluid lactate dehydrogenase 657/serum 191 [U/L], and fluid total protein 39/serum 35 [g/L]).
Chest and abdomen computed tomographic scanning correlated with diagnosis of diffuse lymphangiomatosis, demonstrating widespread smooth interstitial thickening in the lungs with osteolytic lesions in the ribs, and multiple tiny cysts in the spleen (Figure 1). Serum inflammatory markers (such as erythrocyte sedimentation rate, C-reactive protein, and the white blood cell count) were insignificant except for notable increase in the level of serum interleukin-6 (IL-6)reaching up to 946 pg/ml (the normal value is up to 7 pg/ml). Open biopsy from pleural and pericardial tissues confirmed the clinical diagnosis of lymphangiomatosis and excluded other pathological conditions such as malignancy, tuberculosis, or granulomatous diseases (Figure 2).
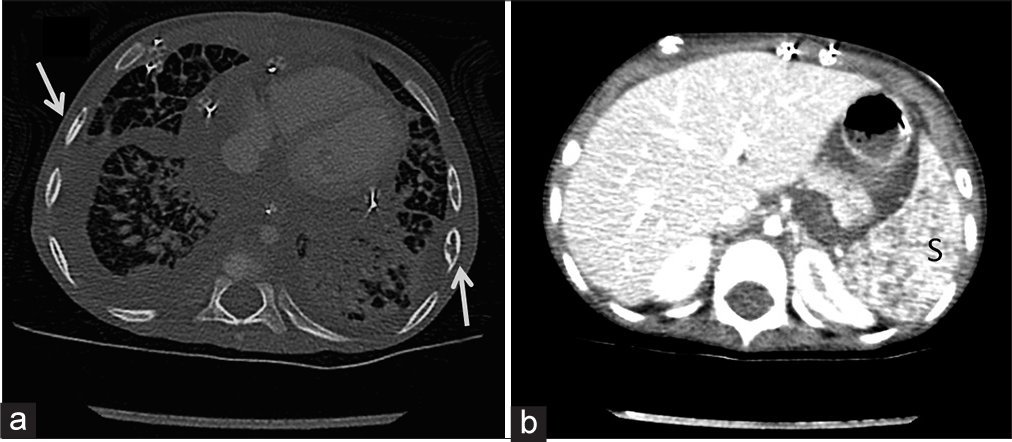
- A 20-month-old boy with diffuse lymphangiomatosis who presented with respiratory failure. (a) Chest computed tomography (CT) with contrast demonstrates diffuse smooth interstitial thickening, multiple patchy air space consolidations mainly in the left; bony lytic lesions involving the ribs bilaterally (arrows); evidence of pericardial effusion and bilateral pleural effusion. (b) Abdomen CT with contrast revealed heterogeneous enhancement of the spleen(s) with multiple tiny cysts.
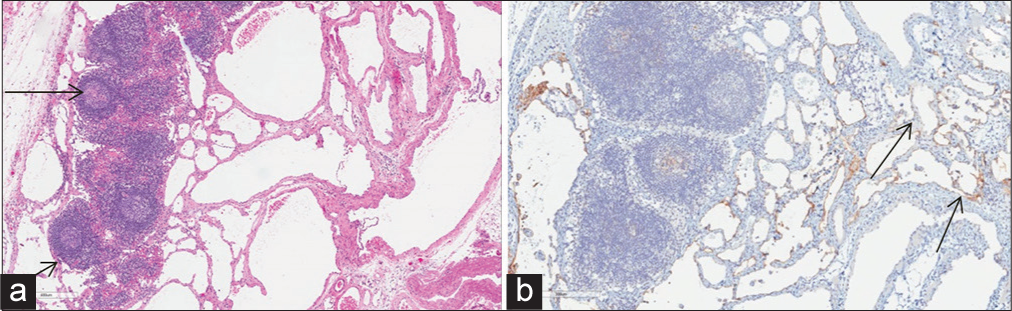
- A 20-month-old boy with diffuse lymphangiomatosis diagnosed by pleural biopsy. (a) Pleura histological section revealed focal reactive hyperplastic lymphoid follicles (arrows) surrounded by lymphangiectatic vessels with the characteristic lymphangitic distribution of abnormally dilated lymphatic spaces. (b) The immunohistochemical staining revealed abnormal lymphatic channels (arrows) positive for the lymphatic endothelial antigen recognized by D2-40 (podoplanin).
The pleural and pericardial fluid output continued to be significant, with mean daily drainage of 1050 ± 158 ml with maximum amount reached up to 6.8 L/day in spite of multiple approaches of conservative and medical management that includes bowel rest with nil per mouth, total parenteral nutrition (TPN), prednisolone, somatostatin agonist (octreotide), and beta-blocker (propranolol).
Due to significant incessant thoracic drainage that frequently affected hemodynamics and respiratory status, a palliative pleuroperitoneal shunt was implanted with creation of pericardial window aiming to direct pleural and pericardial fluid drainage toward peritoneal cavity and to prevent cardiac tamponade. In spite of all previous conservative medical and surgical interventions, the drainage output from chest and mediastinum tubes remained significant requiring meticulous fluid resuscitation and tedious electrolytes replacement. With this refractory chylorrhea, a trial of pegylated interferon alpha-2b (30 µg subcutaneously) was introduced. The patient spiked high fever and rigors immediately after interferon treatment for 3 days that was managed with antipyretics and cold compresses. Following interferon administration, drainage declined precipitously within 2 weeks with no further fluid accumulation in pleural and pericardial spaces (Graph 1). The follow-up of IL-6 showed declined in the level to normal value. The patient was discharged home in stable condition and remained with a satisfactory respiratory status for 6 months, with consideration for further management based on routine follow-up evaluation.
DISCUSSION
Lymphangiomatosis is a rare disease believed to be congenital with no sex preference.[1,2] It has been postulated that the disease results from abnormal lymphatic development.[1,2]
The majority of cases are diagnosed in childhood. It typically manifests as a diffuse lung disease with osteolytic bone lesions. Spleen and liver can also be affected in this disease.[1-3] Lung involvement is a leading cause of death.[1] The osteolytic lesions have been described in the maxilla, mandible, clavicle, ribs, cervical vertebrae, pelvis, and femur.[2] The coexistence of lytic bone lesions and chylous effusion is an important diagnostic clue that facilitates diagnosis but worsens the prognosis.[2] The clinical presentation and radiographic findings may assist in supporting diagnosis, but the final diagnosis is made histologically.[1-4]
In our case, there was extensive chest involvement with diffuse interstitial lung thickening, multiple osteolytic rib lesions, chylothorax, and chylopericardium, in addition to spleen involvement with multiple cystic lesions (Graph 1 and Figure 1). The pleural and pericardial fluid in our patient showed analysis similar to chyle which was defines in literature if fluid triglyceride >1.1 mmol/L and the absolute white cell count >1000/mm3 with dominant lymphocytosis more than 80%.[5] Our radiological findings (Figure 1) correlated with the diagnosis of diffuse lymphangiomatosis as in previously reported cases.[2,3] The diagnosis was confirmed with the characteristic pathological pleural changes (Figure 2)described in previously reported cases.[4]
Chylothorax and chylopericardium are rare conditions seen in infants and children; the etiologies include traumatic and surgical injury to thoracic duct, high systemic venous pressure, idiopathic, malignancy, tuberculosis, and other disorders.[6]
Due to the rarity of lymphangiomatosis, there is no established standard treatment.[1] Conservative treatment options such as TPN, medium chain triglyceride, and high-protein diet with somatostatin agonist (octreotide) have been tried and appear to be frequently ineffective.[7] Recently, it has been shown that propranolol, a non-selective beta-blocker, and steroids may play a role in the treatment of diffuse lymphangiomatosis and in reduction of the amount of chylous effusions.[1,7,8] Attempts and trial of bowel rest, steroid, octreotide, and propranolol in our case did not decrease chyle production. Other options described in multiple case reports with individual success include sclerotherapy with doxycycline, pleurectomy, radiation, chemotherapy, and embolization of lesions.[1,7,8]
Several authors have suggested ligation of thoracic duct and excision of the pathologic lymphangiomatous lesions as a treatment option.[1,7,8] However, this treatment failed in our case, as the radioisotope lymphangiography defined neither ductal structures that can be ligated nor a distinct lymphangiomatous lesion to be excised.
The rationale for pleuroperitoneal shunt with creation of a pericardial window in our case was based on previous published reports[6] to control the chylorrhea in refractory chylothorax and chylopericardium by ensuring adequate drainage and preventing significant pleural collection, pericardial tamponade, and constrictive pericarditis. Sclerotherapy with doxycycline was not tried in our case as it can cause difficulty with drainage of septal fibrous structures after placement of the shunt.
More recently, pegylated interferon alfa-2b therapy has been tested in lymphangiomatosis with significant improvement, although there have been reports of unfavorable outcomes.[1,7] The mechanism of action of interferon is linked to the suppression of tumor cell division and boosting the immune system.[1,7] In general, all reported side effects of pegylated interferon alfa-2b are tolerable and not life-threatening.[1,7] With the incessant pleural and pericardial drainage, we initiated pegylated interferon alfa-2b as adjunct treatment with remarkable improvement in the amount of drainage and overall clinical condition shortly after starting the regimen. No significant toxicity has been observed.
The serum IL-6 level was markedly elevated before treatment with pegylated interferon alfa-2b reaching up to 946 pg/ml (the normal is up to 7 pg/ml). There are few published reports about strong association between splenic lymphangiomatosis and IL-6 production,[2] and it was used as a marker of response to treatment with pegylated interferon alfa-2b.[9] The follow-up of IL-6 level after pegylated interferon in our case showed declined in the level to normal value of 4.9 pg/ml.
Considering lymphangiomatosis as a part of Gorham-Stout syndrome (vanishing bone disease) in our patient is also a possibility that we entertained, especially with some reports suggesting that lymphangiomatosis, and Gorham-Stout disease should not be considered as separate entities, but rather as a spectrum of single disease.[10]
Although our patient was discharged home on room air within 2 weeks of initiating pegylated interferon alfa-2b regimen and remained in a stable condition for 6 months, the prognosis of our case is undefined with possibility of future relapse, reaccumulation of chylous effusion, and even involvement of other organs. Periodic screening with echocardiography, chest X-ray, abdominal ultrasound, and skeletal survey is warranted. Further, management is based on follow-up and ability to sustain good clinical condition with remission of symptoms. Pegylated interferon alfa-2b therapy and radiation therapy have been documented recently to produce temporary or long-lasting symptomatic relief with favorable relative outcome in some case reports.[1,7,8]
In conclusion, the management of chylothorax and chylopericardium in the presence of lymphangiomatosis is controversial and it is medical challenge needs multidisciplinary approaches. A combination trial of surgical intervention such as pleuroperitoneal shunt with a pericardial window creation, and pegylated interferon alfa-2b as adjunct treatment in the presence of high IL-6 was helpful in our case. Generalization of this management requires further studies.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Clinical and radiological features of generalised lymphangiomatosis. Hong Kong Med J. 2008;14:402-4.
- [Google Scholar]
- Splenic lymphangiomatosis with inflammatory signs and elevated serum interleukin-6. J Clin Exp Hematopathol. 2004;44:77-80.
- [CrossRef] [Google Scholar]
- Pathology analysis of a rare case of diffuse pulmonary lymphangiomatosis. Ann Transl Med. 2016;4:114.
- [CrossRef] [PubMed] [Google Scholar]
- Presentations and management of different causes of chylothorax in children: One medical center’s experience. Biomedicine (Taipei). 2017;7:5.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic chylopericardium: An unusual cause of cardiac tamponade. Tex Heart Inst J. 2006;33:249-52.
- [Google Scholar]
- Improvement of disseminated lymphangiomatosis with recombinant interferon therapy. Pediatr Pulmonol. 2000;29:321-4.
- [CrossRef] [Google Scholar]
- Propranolol for intractable diffuse lymphangiomatosis. N Engl J Med. 2011;364:1380-2.
- [CrossRef] [PubMed] [Google Scholar]
- Serum interleukin-6 levels correlate with resistance to treatment of chronic hepatitis C infection with pegylated-interferon-α2b plus ribavirin. Antivir Ther. 2011;16:1081-91.
- [CrossRef] [PubMed] [Google Scholar]
- Angiomatosis of bone and soft tissue: A spectrum of disease from diffuse lymphangiomatosis to vanishing bone disease in young patients. Clin Radiol. 2001;56:184-90.
- [CrossRef] [PubMed] [Google Scholar]




